
Launched in 1994, Deerfield Management Company is an investment firm dedicated to advancing healthcare through information, investment, and philanthropy—all toward the end goal of cures for disease, improved quality of life, and reduced cost of care.
Read More
Investment
Supporting companies across the healthcare ecosystem with flexible funding models…
Read More



Portfolio Companies
Deerfield generally maintains a combined portfolio of more than 150 private and public investments across the life science, medical device, diagnostic, digital health and health service industries at all stages of evolution from start-up to mature company.
Read More View Portfolio Companies
Research Collaborations
Deerfield partners with leading academic research centers, providing critical funding and expertise to further sustain and accelerate the commercialization of discoveries toward meaningful societal impact by advancing cures for disease.
Read More View Research Collaborations
Strategic Partners
As a strategic partner, Deerfield offers capital, scientific expertise, business operating support, and unique access to innovation.
Read More
Deerfield Foundation
The Deerfield Foundation is a New York City-based not-for-profit organization whose mission is to improve health, accelerate innovation and promote human equity.
Read More Meet the Foundation TeamFoundation Partners by Focus
Children's Health Diversity Initiatives Maternal Health View AllFounded Diversity Programs
Break into the Boardroom Fellowships Science to the Street (formerly Women in Science)
Cure Campus
Cure is a 12-story innovation campus in New York City that intends to bring together innovators from academia, government, industry, and the not-for-profit sectors to advance human health and accelerate the fight against disease.
Read More Join the Cure Email ListCure Programming
Cure has a series of expert lectures intended to advance thought in healthcare, management, innovation, policy, and other relevant subjects. This fosters growth and education for those at Cure and its guests.
Events at the Cure
The Need to Expand Buprenorphine Prescribing to Treat Opioid Use Disorder

Overdose Crisis Endures, Yet Few People Receive Existing Effective Treatment
Even before Covid-19, despite enormous unmet need, life-saving buprenorphine is vastly underprescribed
(New York, NY, August 24, 2020) –Approximately half of clinicians authorized to prescribe buprenorphine for opioid use disorder (OUD), one of three medications for OUD, are actively prescribing the medication, according to a JAMA study led by researchers at The Pew Charitable Trusts, Deerfield Management Company, and RAND Corporation.
The research letter, which appeared in the August 24th online issue of JAMA Network Open, examines national opioid use disorder buprenorphine prescribing patterns by Drug Enforcement Administration (DEA)-approved patient limits.
Buprenorphine is considered one of the most effective therapies to treat OUD. In order to become an authorized prescriber of buprenorphine to treat OUD, clinicians are required to undergo special training and licensing with the Substance Abuse and Mental Health Services Administration (SAMHSA) and DEA.
The authors of the paper point out that federal regulations currently limit these waivered clinicians to treating 30, 100, or 275 patients concurrently, with clinicians limited to treating 30 or 100 patients able to request an increased limit.
Based on a national analysis of clinician databases from the DEA and SAMHSA and clinician-level prescribing information from Symphony Health, the authors found that of 55,938 waivered clinicians, only 50.9 percent wrote at least one buprenorphine prescription during the 22-month period of April 2017 through January 2019.
Median patient monthly census calculations revealed 275-patient clinicians treated 36.9% of their patient limit, while 100-patient and 30-patient clinicians treated 23.9% and 11.3% of their patient limits, respectively.
“More than 2 million people in the United States have an opioid use disorder, yet few of them receive any type of specialty treatment, including buprenorphine,” said study co-author Alexandra Duncan, senior officer with The Pew Charitable Trusts’ substance use prevention and treatment initiative. “Removing barriers to buprenorphine prescribing can help close this treatment gap and ensure that people have access to the evidence-based addiction care they need.”
In recognition that buprenorphine is an important option for patients because it eliminates the need for the daily clinic visit required of most patients receiving methadone, NYC Health and Hospitals expanded access to the medication by integrating prescribing into primary care. Additional benefits of buprenorphine are its low potential for abuse and negligible risk for overdose.
“Our finding that about half of doctors who can prescribe buprenorphine aren’t doing so makes clear that increasing the number of patients receiving it is not just about increasing the number of clinicians who can prescribe it,” added co-author Bradley Stein, Director of RAND’s Opioid Policy Center. “We need to focus efforts on increasing reimbursement for buprenorphine’s use, educating prescribers, patients, and their families about its effectiveness, and combatting the stigma that hampers the effective treatment of opioid use disorder.”
“Leveraging multiple large databases not only allowed us to confirm earlier evidence that clinicians are prescribing below their patient limits, but also enabled us to continue to peel back the onion on the scope of this problem, said co-author Jared Anderman, director of data analytics at the Deerfield Institute, a division of Deerfield Management Company. “We are looking forward to continuing to work with the powerful dataset we have created and identifying additional opportunities to make an impact.”
Authors of the study, titled, “Monthly Patient Volumes of Buprenorphine-Waivered Clinicians in the U.S.,” are: Alexandra Duncan (The Pew Charitable Trusts); Jared Anderman (Deerfield Management Company); Travis Deseran (previously Deerfield Management Company); Ian Reynolds (The Pew Charitable Trusts); and Bradley D. Stein (RAND Corporation). The work was funded by The Pew Charitable Trusts and Deerfield Management Company.
-####-
About Deerfield Management Company
Deerfield is a healthcare investment management firm committed to advancing healthcare through investment, information and philanthropy.
Media Contacts:
Karen Heidelberger, [email protected], 212-692-7140
Warren Robak, [email protected], 310-451-6913
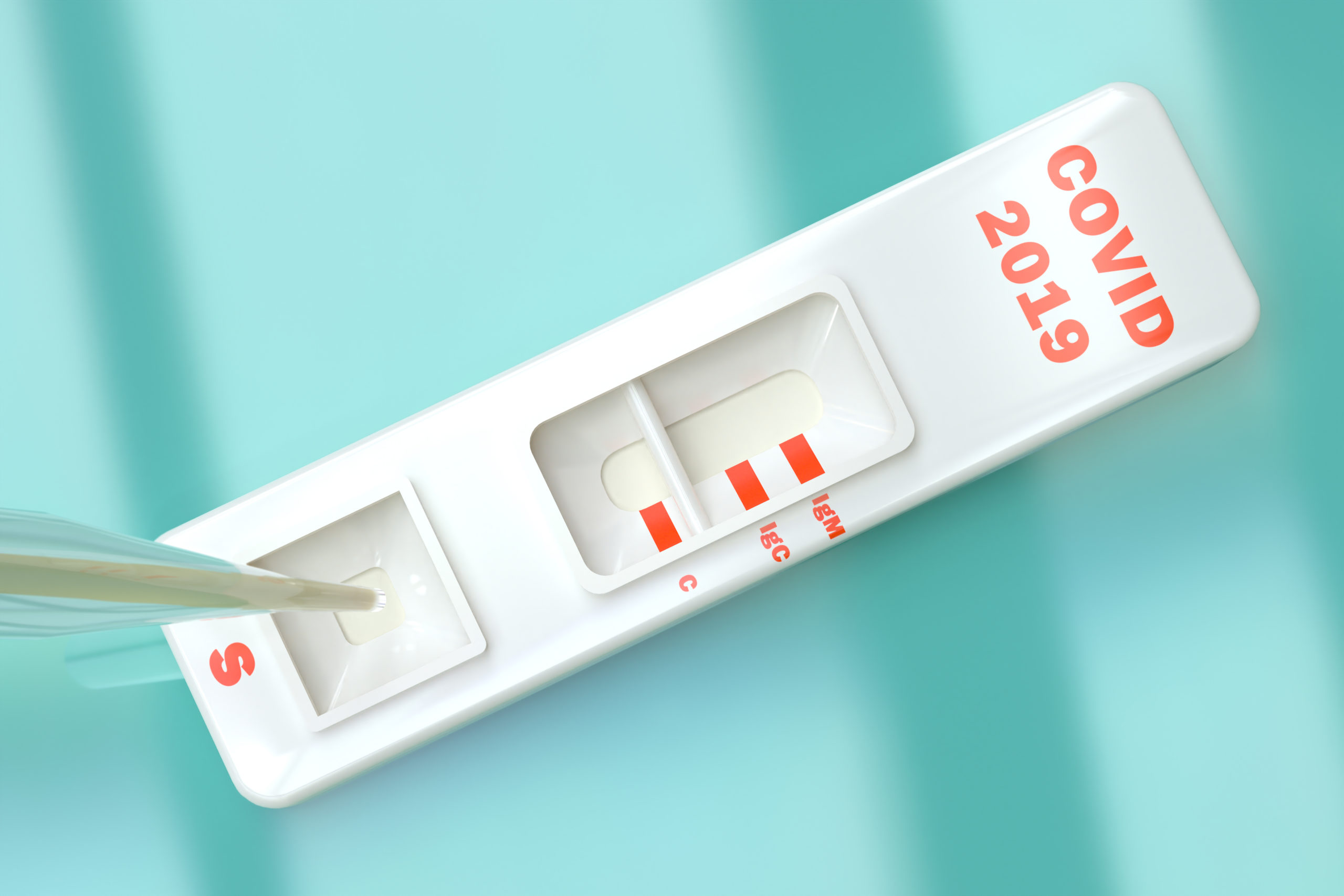
Deerfielders Weigh in on a Safe Return to Work Policy Amid Covid-19 Crisis
Antibody testing provides a data-driven path to getting people back into the economy
The availability of point of care antibody testing—also known as serological testing—may provide a feasible roadmap for getting people back to work safely following the COVID-19 crisis, according to an editorial published in the journal Contemporary Clinical Trials Communications.
“You can’t stop the economy forever,” asserted Governor Cuomo in a recent news conference, according to STAT. “So we have to start to think about, does everyone stay out of work? Should young people go back to work sooner? Can we test for those who had the virus, resolved, and are now immune, and can they start to go back to work?”
Regardless of whether they already have immunity to the virus, millions of Americans may try to return to work, potentially undoing all the benefits of the shutdown, suggests the editorial.
Antibody testing, the authors argue, could clarify a person’s status quickly in real-time and reveal whether they have been exposed to COVID-19. Accordingly, a person who mounts an IgG positive response (suggesting the presence of immunoglobulin G antibodies) would most likely now be immune to the virus and an IgM positive result would point to the process of developing immunity in someone who more recently became infected.
“Unlike the PCR tests (a measure of virus material), the immediate results and unconstrained supply of antibody tests could fundamentally change the way we manage this epidemic,” says Robert Jackson, MD, a co-author of the paper. “And from an economic perspective, it could lead to a tractable path for people to return to work. Collecting the data and tracking individuals longitudinally, in order to confirm the hypothesis, will be necessary.”
And barring any HIPAA concerns, the authors propose that persons with positive antibody tests during periods of social distancing could get a bracelet, which indicates that they are immune-protected and can return to work. Those without a bracelet would still be asked to practice social-distancing and not yet resume their normal activities. But this approach could potentially get at least some portion of the economy back running again, suggests the authors.
According to the authors, the antibody tests are cheap, easy to administer, and could be made available at every hospital.
“Broad testing is in society’s best interest,” says Alex Karnal, a co-author of the editorial. “Until we make serological tests available in a robust way, it’s as if we are flying a plane without navigation.”
Authors of the editorial, titled, “Let’s Get Americans Back to Work Again,” are: Alex Karnal, Partner and Managing Director; Robert Jackson, MD, Partner and Chief Science Officer; and Joe Pearlberg, MD, PhD, Vice President of Scientific Affairs, all at Deerfield; and Amitabh Chandra, PhD, McCance Family Professor at Harvard Business School and Weiner Professor at the Harvard Kennedy School.

Deerfield Contributes Insights to Peer-Reviewed Study on Access to Life-Saving Drug, Buprenorphine, Examining Growth and Distribution of Waivered Providers
Despite Evidence Showing the Opioid Crisis Disproportionately Affects Rural Areas, Prescriber Growth There Remains Considerably Slower
Despite Evidence Showing the Opioid Crisis Disproportionately Affects Rural Areas, Prescriber Growth There Remains Considerably Slower
While there has been an uptick in the number of U.S. clinicians having waivers to prescribe the potentially life-saving drug, buprenorphine, the total number of waivered prescribers in 2017 still represented fewer than 10 percent of all primary care providers, found a report published online in the January 7 issue of the Annals of Internal Medicine.
Moreover, although rural communities have been shown to be disproportionately affected by the opioid epidemic, the growth in the number of providers having this required certification there remains strikingly low, compared to more urban areas. Authors from the RAND Corporation suggest a need for more targeted efforts to increase access to the medication.
To assess the growth in buprenorphine-waivered providers by region and demographics, the investigators leveraged insights from analysis performed by the Deerfield Institute.
Tapping population estimates from the 2010 U.S. census and total physicians per capita, the researchers calculated the total number of waivered providers per 100,000 from 2007 to 2017. Statistics from the Census Bureau were also used to determine per-capita sociodemographic characteristics.
Over the decade studied, the researchers found that the number of waivered providers, in general, increased from 3.80 to 17.29 per 100,000 persons. Growth rate of waivered providers was markedly slower in small, nonmetropolitan areas, as it was in communities with lower levels of education.
The Food and Drug Administration approved buprenorphine for treating opioid dependency in 2002. According to Kaiser Health News, once physicians secure the waiver, they can prescribe buprenorphine in a range of settings, including primary care offices, community hospitals and correctional facilities. Compared with methadone, Buprenorphine is less likely to result in fatal overdoses.
The federal government is undertaking a number of efforts to increase the amount of buprenorphine prescribers.
More details on the research may be found here: Annals of Internal Medicine.
The authors of the paper are Ryan K. McBain, PhD, MPH, Andrew Dick, PhD of the RAND Corporation in Boston, Massachusetts and Mark Sorbero, MS, and Bradley D. Stein, MD, PhD of the RAND Corporation in Pittsburgh, Pennsylvania.

Generic drug prices are actually falling
Despite drop, patients continue to get hit with high prices at the pharmacy for reasons that may have little to do with pharma
As pharma gets slammed for egregious pricing of life-saving medications and stories continue to make headlines – generic drug prices are actually falling.
That’s not to say that consumers aren’t still being saddled with high costs at the pharmacy, but a July 2019 paper by the National Bureau of Economic Research (NBER) that reviewed the pricing patterns of generic drugs suggests that other factors may be at play.
Although the U.S. generic prescription pharmaceutical market continues to drive overall prices downward, reductions in pharmacy price are not fully passed to patients, according to the NBER researchers.
One contributing factor to patients not reaping as much of the benefit of the generic price declines, the authors suggest, is the increase in cost-sharing: the practice of insurers offering plans that increasingly shift costs from insurers to consumers.
“Plan sponsors are opting for benefit designs that have consumers sharing a higher percentage of costs. The result is out-of-pocket costs falling less than overall generic drug prices,” said Deerfielder Vince Mellet. “Even so, the prices of generic drugs went down.”
To put these generic price increases into context, the investigators developed two price indices that capture prices of generic prescription drugs paid by consumers of private health insurance plans.
The first, direct out-of-pocket CPI, measures consumers’ direct out-of-pocket payments to the dispensing pharmacy. The second, total CPI, represents the total revenues received by the dispensing pharmacy – the consumers’ direct out-of-pocket payments, plus the amount paid to the pharmacy by the insurer on behalf of the customer.
Based on the analysis, the researchers found direct out-of-pocket CPI for generic prescription drugs declined by approximately 50 percent between 2007 and 2016, while the total CPI fell by nearly 80 percent over the same period. The investigators partly attribute the smaller reduction in the direct out-of-pocket CPI, compared to the total CPI, to consumers’ increasingly moving away from fixed copayment benefit plans to exclusively coinsurance or a mix of coinsurance and copayments.
While consumers are encountering more cost-sharing that shifts more of the drug cost burden on to them, the researchers report that, on balance in the U.S., consumers have still experienced significant price declines for generic drugs.
Given their findings, the investigators suggest that overall affordability is not the primary issue in the generic drug market and that this segment of the U.S. prescription market is not responsible for the reported growth in prices and spending for prescription drugs overall.
To get the full picture on prices in the U.S. generic prescription industry, the NBER researchers recommend taking a closer look at all components of the entire generic supply chain, from manufacturer, wholesaler, pharmaceutical benefit manager, insurer, to retailer.
To view the full paper, titled “The Price to Consumers of Generic Pharmaceuticals: Beyond the Headlines”, visit: https://www.nber.org/papers/w26120
© 2019 by Richard G. Frank, Andrew Hicks, and Ernst R. Berndt.

Revisiting Biosimilars: A Closer Look At The Commercial Barriers
In a past issue of this newsletter (September 2017) we took a broad look at the salient issues pertaining to the realm of biosimilars, including regulatory, legal, and commercial aspects of the debate. We refer readers here for an initial grounding if needed. In the intervening time period, new developments have played out that highlight the significant barriers to commercial uptake that exist for these products.
Briefly, biosimilars are to biologics as branded drugs are to generics. However, both biologic drugs and biosimilars are by and large exceedingly more difficult to manufacture and thus bring to market as the production process is critical to the potency and release quality of these drugs within parameters established by the innovator product. There are only yet 15 biosimilars approved in the US, and only five are actually commercially available, with the remainder being still blocked from the market due to patent exclusivity. Contrast this to Europe, where 40 biosimilar products are approved.
The cost of production for these products is not nearly at the level of generics. Take insulin, for example. Insulin has a somewhat hybrid role in the US, as the first formulations were approved before the biologics approval pathway existed in the US, so its “biosimilars” are approved under what is known as the 505(b)2 pathway in the US, as opposed to the 351(k) pathway used by products whose innovator products are a true biologic (as determined by approval pathway). The market would seem ripe for a Lantus biosimilar, as the product has greater than $4bn in annual sales. Eli Lilly already has a biosimilar to Lantus on the US market, called Basaglar, which is annualizing greater than $600m through its first six quarters into launch.
Merck, through a collaboration with Samsung Bioepis had a tentatively approved biosimilar to Lantus from July 2017. However, a filing by Samsung Bioepis in October 2018 indicated Merck had cancelled development and commercialization contracts for the product. One analyst report cited the decision to pull out as being due to a review of the market environment and production costs of insulin biosimilars. If a biosimilar as relatively “simple” as insulin can be difficult to manufacture at scale with attractive margins, then this certainly cannot bode well for more complex protein products to do the same. With Merck’s exit, only Mylan, in collaboration with Biocon, is developing another Lantus biosimilar. While district court litigation is ongoing, a launch for Mylan and Biocon’s product is expected in the 2020-2021 timeframe.
Similarly, Momenta recently announced it would be cutting its biosimilar efforts altogether, and simultaneously cutting half its staff while it refocuses on its non-biosimilar pipeline.
It’s a (rebate) trap
FDA Commissioner Scott Gottlieb further shed light on another major commercial obstacle for biosimilars, dubbed the rebate trap, in a March 2018 speech at the America’s Health Insurance Plans National Health Policy Conference[1]. The deep discounts offered on certain branded specialty drugs (often biologic), in the form of rebates and other payment or contractual mechanisms, in many instances can be upwards of 40% to attain preferred formulary positions from pharmacy benefit managers (PBMs) and health insurers. Often these negotiated discounts are volume-based, so the greater the utilization over competitive products, the greater the spread from the wholesale acquisition cost (WAC) to the net price to the plan, with PBMs and health insurers earning a percentage of the spread. Launched biosimilar products have generally been introduced to the market with roughly 15-20% discounts to the WAC of originator products, and thus are unable to displace the originator product. Biosimilars get stuck in a catch-22 situation, where they lack enough patient share for plans to consider moving them to a better formulary position but are hindered from getting more meaningful market share while they sit on a lower formulary tier. PBMs remain financially incentivized to limit the uptake of biosimilars to maintain the flow of rebate payments on originator products. Simply further discounting the biosimilar so that the WAC is on par with the net price of the originator is easier said than done for reasons noted earlier, namely that these drugs have turned out to be much costlier to develop and manufacturer than earlier predictions.
PBMs are attempting to move past the bad press they have received around the rebate trap and the general practice of collecting rebates. Express Scripts recently announced the launch of its Flex Formulary, which will consider authorized generics of branded drugs for inclusion on the formulary, in either a preferred or non-preferred position, and, importantly, discontinue coverage of the branded product. This could foreseeably open the door to more biosimilar adoption on this formulary. The first products added to the Flex Formulary were Epclusa and Harvoni, two hepatitis C drugs made by Gilead Sciences.
Humira in the hotseat
Gottlieb has gone so far as to suggest a competitive bidding scheme for biologics would be ideal[2], which is more akin to the experience in the EU. For example, Remicade sales have fallen over two-thirds in the three years since the introduction of its first biosimilar in the EU. Another closely watched drug is Humira, the world’s top-selling drug, as four biosimilars have just launched in the EU. One analyst report has said AbbVie, the maker of Humira, has won its first tender in Europe by offering an 80% discount off the price prior to the launch of biosimilars[3]. The deep discounting shows the extent to which the company is willing to go to hold onto market share.
Humira biosimilars in the US are still a pipedream, protected by a patent thicket the company has created with additional patents on formulations changes and extending life as new indications are approved. AbbVie has forged confidential legal settlements with several biosimilar makers, that will keep biosimilar copies of Humira at bay until 2023. In the meantime, AbbVie and Amgen are likely to continue to find themselves under increased scrutiny over the practice of repeated price increases for their top-selling drugs. A recent article cited nearly a 140% overall price increase for both of those drugs since January 2013[4]. Without biosimilar competition, and assuming continued price increases on par with recent history, both drug makers could see themselves in the hot seat in the court of public opinion.
[1] https://www.fda.gov/NewsEvents/Speeches/ucm599833.htm
[2] https://www.fda.gov/NewsEvents/Speeches/ucm613452.htm
[3] https://www.statnews.com/pharmalot/2018/11/01/abbvie-humira-biosimilars-prices/
[4] https://www.statnews.com/2018/11/14/humira-abbvie-amgen-enbrel-price-hikes-biosimilars/

New Story Reignites Old Debate On Conflict Of Interest
Drug and device development requires extensive expertise, is costly, and time consuming. In particular, the need for deep expertise in many disciplines means there is a market value for those skill sets, and sometimes those exist in the private sector, but many times, in academia. The relationship between academia and industry is both praised and criticized, but unavoidable as increasingly complex diseases are tackled by both parties. As a result, many academics also have some kind of corporate relationship, be it as a trial investigator, a consulting or advisory role, or a speaker’s bureau slot, to name a few examples. As these relationships merit payment, critics feel industry payments erode away at academics’ ability to offer unbiased opinions and expertise, precipitating the need for some kind of disclosure.
Putting aside the details of the most recent example of a debate over conflict of interest (COI) out of Memorial Sloan Kettering Cancer Center (MSKCC), much of which has already been detailed elsewhere[1], the response suggests disclosure is not only preferred but necessary, though the who, how, and why have yet to be hammered out. Medical journals and some specialty societies may require the data, but do not have uniform criteria for the reporting of industry ties (whether by physicians or sponsors) nor a readily identifiable platform for prospective patients or industry observers to view what relationships physicians may have.
The most comprehensive source of information is the Open Payments database maintained by the Centers for Medicare and Medicaid (CMS).[2] Applicable manufacturers and group purchasing organizations are required to annually submit relevant payment information to physicians and teaching hospitals to CMS, and CMS subsequently allows physicians and those hospitals to review the reported payments for accuracy. Payments for research, meals, travel, gifts, and speaking fees are all required to be reported.
While the most comprehensive, there are some notable exemptions to the Open Payment database, which some argue are loopholes asking to be closed. Companies that do not yet have a marketed product approved by the Food and Drug Administration do not have to report payments, meaning many small biotech companies working on their very first product are not reporting to the database. Additionally, continuing medical education (CME) payments are also exempt, though some argue CME is simply another avenue for industry to market their new launches and pipelines.
Referring to the recent MSKCC news, medical ethicist Dr. Arthur Caplan publicly commented “We have yet to figure out what COI means or how to manage it in a health care world where industry ties are everywhere”[3] which is likely the best succinct encapsulation of the status quo. It is unknown what payment threshold might influence physician decision making (whether perceived or actual), but there is also some intangible value to physicians who serve as investigators on cutting edge clinical trials and serve on advisory boards alongside the top minds in their fields, who in turn can provide better care for their patients. It would be wrong to suggest that all interactions between industry and physicians are either unethical or inappropriate, but we can, and should, do better at reporting them.
[1] https://www.nytimes.com/2018/10/12/health/memorial-sloan-kettering-cancer-disclosure.html
[2] https://openpaymentsdata.cms.gov/
[3] https://twitter.com/ArthurCaplan/status/1038849711174762499

Inaugural Approvals For RNA-Based Medicine
In the case of another historic first in the world of healthcare, this summer saw the first US and EU regulatory approvals for an RNA interference (RNAi) therapeutic, in the form of Alnylam’s Onpattro (patisiran). The drug, given as an infusion, was approved to treat polyneuropathy of hereditary transthyretin-mediated amyloidosis (hATTR) in adult patients. A rare and often fatal disease, hAATR is characterized by the buildup of abnormal amyloid protein in peripheral nerves, the heart, and other organs. Up until this approval, the American College of Cardiology’s recommendations for treatment were merely supportive care and clinical trials, pointing to the level of unmet need for this patient population[1].
Still, it has been anything but smooth sailing for Onpattro, nor for the field of RNA-based medicines. Like gene therapy, it has seen an earlier wave of enthusiasm come crashing down in the wake of technical challenges and safety issues. While Onpattro will not be a panacea to all the known issues, it is important to note where and how it has succeeded where others have failed and to understand what it means for other candidates in the pipeline.
How it works
To understand how RNAi works, transport yourself to your high school biology class where you learned about the “central dogma” theory that explains the relationship between DNA, RNA and proteins – that DNA in the nucleus forms genes which are the template for transcription to RNA, which in turn is the template for translation to proteins. Those proteins then serve as a central player in most biological systems. In individuals without hAATR, the liver produces the TTR protein, used to transport vitamin A and a thyroid-binding protein in the body. Patients with hATTR have a mutation in the gene for TTR, which leads to the creation of a defective and unstable TTR protein. Onpattro binds to the mutated mRNA sequence that causes the defective protein, and cuts out that sequence, effectively halting the production of the misfolded and defective protein[2]. Of note, this approach targets the upstream cause of the defect, and not simply the downstream symptoms that are the manifestation of the defect.
Location, location, location
RNAi does not simply work by targeting the mutated mRNA of interest – it must additionally be specifically targeted to the tissue(s) of interest, which is the tougher nut to crack. The liver, the target organ of interest in hATTR, happens to be better suited for targeted drug delivery given its unique vasculature – notably that it has both the hepatic artery and hepatic vein, allowing it to effectively double dip on whatever has been infused into the blood stream, compared to other organs. It will be more challenging to deliver RNAi to other organs, which thus far has been a challenge to do at therapeutically appropriate levels.
Onpattro is encased in a lipid nanoparticle to help carry the drug to the liver and enter the cell. The lipid nanoparticle disrupts the cell membrane to allow Onpattro in to the cell, and thus can trigger an immune response, so patients must prophylactically take a steroid, acetaminophen, and antihistamines. There is thus also the need to find delivery vehicles that are slightly more sophisticated to obviate any immune responses.
How did we get here?
Pharma entered the RNAi fray in the early 2000s with some big dollar risk-taking in a hot field that would go on to earn a Nobel prize for its scientific discoverers, only to encounter future obstacles. Novartis and Roche paid their way into accessing Alnylam’s platform in 2005 and 2007, respectively. Merck paid $1.1bn to acquire Sirna Therapeutics, Alnylam’s main competitor at the time. Prior to that transaction, Sirna had a research deal with GlaxoSmithKline as well. Early attempts in ophthalmology and other liver targeted attempts encountered issues with RNA degrading before reaching its target, and thus required extremely high dosing to show any efficacy. Novartis and Roche parted ways with Alnylam in 2010, and Merck eventually sold Sirna IP to Alnylam in early 2014 at a massive discount to its earlier purchase. In the interim, Alnylam soldiered on with some restructuring and a deal with Sanofi dating back to 2012 that helped keep it afloat[3].
Looking ahead
Alnylam has said the list price for one year of treatment in the US is $450,000 and has at least one value based agreement, with Harvard Pilgrim Health Care. It plans to officially launch Onpattro later this year and will face competition in the US from Ionis and Akcea’s also newly approved Tegsedi (inotersen), and possibly also Pfizer’s Vyndaqel (tafamidis) before the end of this year.
Alynlam and others are pursuing experimental RNAi treatments in other indications, including acute hepatic porphyrias, cardiovascular disease, hepatitis B, alpha-1 antitrypsin deficiency, primary hyperoxaluria, delayed graft function, and alcohol use disorder[4]. Notably, several of these are also liver-targeted indications with various delivery methods to help with targeting.
[1] http://blogs.sciencemag.org/pipeline/archives/2017/09/20/alnylam-breaks-through
[2] https://www.fda.gov/Drugs/NewsEvents/ucm615953.htm
[3] http://cienp.org.br/wp-content/uploads/2018/03/Alnylam-prepares-to-land-first-RNAi-drug-approval.pdf
[4] https://www.wsj.com/articles/fda-approves-first-drug-based-on-gene-silencing-research-1533923359

Analysis of success rates for the Center for Medicare and Medicaid’s new technology add-on payment program
Objectives
To quantify the approval, denial, and withdrawal rates and identify any predictors of success or failure for all new technology add-on payment (NTAP) applications from FY 2003 to FY 2018 in the United States.
Methods
The Center for Medicare and Medicaid (CMS) releases inpatient payment methodology rulemaking annually in the Federal Register, including details of NTAP submissions. The proposed and final rulemaking documents were analyzed to quantify the approval, denial, and withdrawal rates of all applications and determine primary reasons for denial or withdrawal from FY 2003 to FY 2018. Raw data were coded to further examine any predictors of application success such as product type, therapeutic category, manufacturer type, reapplication status, and proposed rule determination.
Results
There were 95 NTAP applications submitted over the last 15 fiscal years. Approximately 30%, 25%, and 45% of applications were approved, withdrawn prior to final rule, or denied, respectively. Inability to meet the “newness criteria” developed by CMS was the primary reason for denied and withdrawn applications. Product type, therapeutic category, and reapplication status have minor to significant impact on the approval rate of an application. However, manufacturer type and proposed rule determination have little to no impact on application outcome.
Conclusions
While there are a few factors that may positively influence the outcome of a NTAP application, the approval rates for the program are low overall. Without additional reimbursement from the NTAP program, inpatient hospitals may be deterred from adopting innovative therapies because of financial burdens. CMS and manufacturers should strive to find a better consensus for a framework that adequately incentivizes the utilization of new technologies for Medicare beneficiaries.

Labs Recalibrate To New Normal Under Pama Prices
As of January 1st of this year, the rate-setting scheme for nation-wide payment rates from the Centers for Medicare & Medicaid Services (CMS) has been rejiggered in an attempt at allowing CMS to benefit from the type of rate contracting that private payers have long been able to do. While CMS has historically set rates for laboratory tests, with private payers then typically using that price as a starting point for negotiation, the new system, as established by the Protecting Access to Medicare Act of 2014 (PAMA), flips the roles of the two, and instead uses the private-payer rates to set Medicare pricing. PAMA is one attempt to cut CMS in on the action and allow it to benefit from free-market based negotiations that private payers do as a natural course of business.
Despite the four-year lead time from the passage of PAMA to implementation of the rule (which was even one year later than it was supposed to be, by statute), there were still surprises once the final rates were released, and perhaps more importantly, leaves questions about what the new status quo could and/or should be with respect to the elaborate choreography of rate setting, negotiating, and contracting.
Under PAMA, labs are required to report to CMS the private payer rates they pay for particular tests. CMS then determines a weighted median amount by test in order to set the new prices. This is not a one-time submission – labs will be required to continue submitting payment data in future years. However, rates cannot be reduced by more than 10% each year for the first few years, and no more than a 15% reduction in the following few years, as a means of setting a floor on pricing expectations to protect labs. CMS has by and large been excluded from negotiating with manufacturers of all kinds on cost, and instead has had to accept list prices across the board.
One of the biggest criticisms around the implementation of PAMA is the criteria used to determine exactly which labs are meant to submit their data to CMS. One estimate has only 34% of the lab market represented, with two major labs representing 80% of the volume used to calculate the rates[1]. Labs are required to report their data if over 50% of their revenues are from Medicare, though, with a bit of handwaving, manages to leave hospital-based labs out of those required to report. Once the hospital lab costs are taken altogether with all other costs of the hospital, including lucrative inpatient stays for privately insured patients, they end up falling under that 50% threshold. It has been suspected hospital labs have been paid somewhat higher than commercial labs (the Quest Diagnostics and LabCorp’s of the world), so leaving hospital labs out of the mix has a downward effect on the median price calculation.
It is also worth noting PAMA regulations do come with some teeth: steep monetary fines can be levied against any lab that is required to report but does not. Though because criticism has more been around erroneously leaving some labs out rather than incorrectly bringing too many labs into the fold, it is not likely these types of fines will often come into play. There is a minimum expenditure threshold for labs of $12,500 of Medicare revenues from the clinical lab fee schedule (CLFS) as well, and so probably correctly does not include those outlier labs with low overall total Medicare payment dollars. The CLFS is the nationally set standard for what Medicare will pay for outpatient clinical lab services.
All of these changes work out to pretty significant dollars – CMS pays about $7bn per year for clinical lab tests. In fiscal year 2018, CMS expects savings close to $400m dollars, and up to $1.7bn and $3.9bn in savings over the next 5 and 10 years, respectively[2]. The top 20 codes with the greatest reductions in payment rates all saw cuts greater than 59%, with one code payment rate even cut 99.99%.
Still, some tests saw significant increases in their payment rates. The top 20 largest increases for certain codes range from a 150% increase all the way up to a 750% increase (looking at you, “urine screen for bacteria”!)[3].
There are still some implications to PAMA that have fully yet to be felt. Notably, the Medicare CLFS rates were often seen as the starting point for contracting negotiations between labs and payers, and likely arriving at a lower rate than the CLFS fee schedule for private payers – i.e., payment rates defined broadly as a 20% haircut from CLFS rates. Large private payers like the Blue Cross Blue Shield plans have been known to recalibrate their negotiated rates every 3-5 years off the CLFS. But if private payers are looking for a discounted rate off the CLFS fee schedule, and then labs subsequently submit those rates to CMS for the PAMA recalibration of CLFS rates, it quickly becomes a race to the bottom in terms of dollars flowing to labs.
The American Clinical Laboratory Association (ACLA), a large trade group representing labs, has even gone so far to file a lawsuit challenging the PAMA reimbursement rates[4]. As of the time of writing, this suit is still ongoing and has yet to be resolved or a judgement issued. The intricate choreography of negotiating and rate setting between private payers, labs, and CMS will also clearly need to be re-thought. This early on, nothing is probably yet off the table, and does open some opportunity for new and creative schemes to come into play in an arena where we and others will continue to watch.
[1] https://www.xifin.com/news/press-releases/2017/xifin-highlights-flaws-cms-2018-draft-pama-pricing
[2] https://www.genomeweb.com/molecular-diagnostics/cms-holding-market-based-payment-labs-until-2018-tweaks-criteria-labs
[3] https://www.xifin.com/resources/blog/201803/understanding-pama-changes-and-managing-its-effects
[4] http://www.acla.com/cms-ignored-congressional-intent-in-implementing-new-clinical-lab-payment-system-under-pama-acla-charges-in-suit/